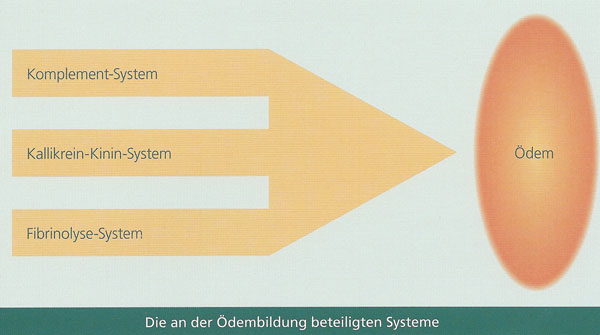
L’angio-œdème héréditaire (AOH) est une maladie rare, transmise sur le mode autosomique dominant et causée par un défaut génétique d’une protéine qui entraîne une dysrégulation de différents systèmes, par exemple du système du complément, du système des kinines, du système de la coagulation ou du système fibrinolytique. Il s’agit donc d’un facteur à fonctions multiples. Ce facteur s’appelle inhibiteur de la C1-estérase (C1-INH). Le déficit en C1-INH (AOH de type I) ou la déficience fonctionnelle de C1-INH (AOH de type 2) se traduisent par une activation incontrôlée des systèmes mentionnés plus haut. Cela favorise la formation d’une substance (bradykinine) qui a pour effet d’augmenter la perméabilité des vaisseaux sanguins (perméabilité vasculaire) et par voie de conséquence de provoquer un épanchement de liquide dans les tissus (œdème). Etant donné que chez les patients AOH l’œdème est dû à la défaillance d’un système extrêmement complexe, on comprend pourquoi les interactions entre les différents systèmes impliqués et leurs systèmes de régulation n’ont pas encore été complètement élucidées.
Types d’AOH
On distingue deux types d’angio-œdème héréditaire :
Type I
L’AOH de type I est la forme la plus fréquente (85 % des cas connus). Le type I se caractérise par un déficit partiel ou complet en inhibiteur de la C1-estérase.
Type II
Dans le type II, la concentration de C1-INH est normale mais l’activité de la molécule est diminuée, voire inexistante.
AAE 1
L’angio-œdème acquis (AOA) est encore plus rare que l’AOH. Dans le cas présent, l’AOA est associé à une autre maladie qui doit être impérativement élucidée. Etant donné que l’AOA n’est pas héréditaire, il n’y pas d’anamnèse familiale.
AAE 2
L’AOA de type 2 est une réaction auto-immune au cours de laquelle des anticorps suppriment la fonction de l’inactivateur C1.